ABINIT-MP Openシリーズ (Ver.2 Rev.8)
※2021年9月版(Ver.2 Rev.4)に関するページはこちらです
※2020年6月版(Ver.1 Rev.22)に対応するBSV(BioStation Viewer)のWindows用インストーラはこちらからダウンロードできます
はじめに
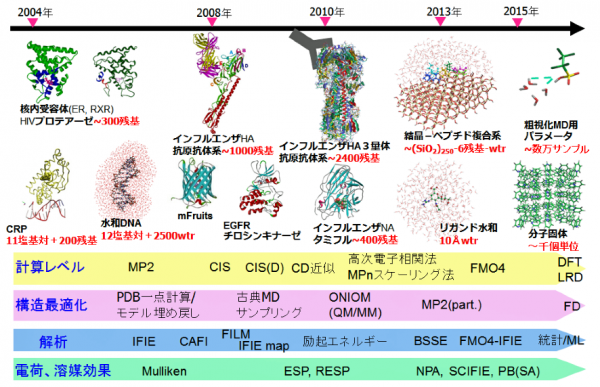
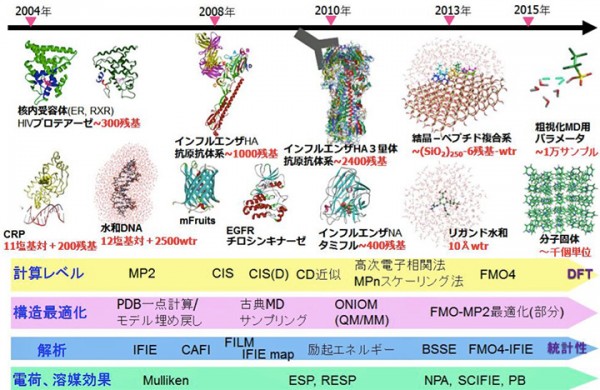
ABINIT-MP [1,2]のOpenシリーズはVer. 2系に移行し、初回のリリースが2021年9月のRev. 4 [3]、今回が2回目のRev. 8 [4]となります。Rev. 4の頁でも記していますが、Ver. 2系では機能の拡充と共に、常用されるFMO-MP2ジョブの高速化、さらに数万フラグメントの扱いを目指した整備が行われています。高速化については、計算機科学の高性能計算(HPC)の専門家グループとのコラボレーションの下、「富岳」に代表される富士通A64FX系のスパコンを意識しての改良作業となっています。2020年6月にリリースしたVer. 1系の最終版Rev. 22に比べるとFMO-MP2計算は、前回のVer. 2 Rev. 4では1.2~1.4倍の加速、今回のVer. 2 Rev. 8では1.5~2倍の加速となっています。大規模系への対応に関しては、タンパク質の水和モデルの扱いにおいて遠方の水分子をクラスターとしてまとめ、フラグメント総数を実効的に削減する前処理スクリプトを開発しました。これにより、新型コロナウイルスのスパイクタンパク質の扱いが可能となりました。
機能面での強化では、PIEDAで局所応答分散(LRD)法によって分散力の寄与を電子相関の寄与と分離して評価できるようになった他、興味領域での励起エネルギーとイオン化エネルギーの算定が可能となりました。また、分子凝集系を想定した励起状態でのフラグメント間での結合パラメータを評価する機能も追加されました。
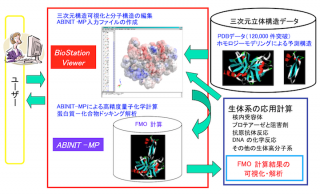
Ver. 2系ではCPF(Check Point File)の書き出しが無効化されています。CPFの読み込みによるBSV(BioStation Viewer)を使った可視化的な解析については、今後も当面並行して提供するVer. 1 Rev. 22を使っていただくことになります。「富岳」を頂点とする国内HPCI拠点では、Ver. 2系の更新版と共にVer. 1 Rev. 22もライブラリとして利用できるようにしています。
Ver. 2 Rev. 8では、2つのネームリスト(&EXPROPと&COUPLING)が追加されており、Ver. 1 Rev. 22とVer. 2 Rev. 4のajfファイルに追加しないと読み込みに失敗します。そのため、mkinp_ver2rev8-hd-2023august.py スクリプトを使ってVer. 2 Rev. 8用に転換する必要があります。
[1] “Electron-correlated fragment-molecular-orbital calculations for biomolecular and nano systems”, S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama, K. Fukuzawa, Phys. Chem. Chem. Phys. 16 (2014) 10310-10344.
[2] “The ABINIT-MP Program”, Y. Mochizuki, T. Nakano, K. Sakakura, Y. Okiyama, H. Watanabe, K. Kato, Y. Akinaga, S. Sato, J. Yamamoto, K. Yamashita, T. Murase, T. Ishikawa, Y. Komeiji, Y. Kato, N. Watanabe, T. Tsukamoto, H. Mori, K. Okuwaki, S. Tanaka, A. Kato, C. Watanabe, K. Fukuzawa (pp.53-67) in “Recent Advances of the Fragment Molecular Orbital Method – Enhanced Performance and Applicability”, ed. by Y. Mochizuki, S. Tanaka, K. Fukuzawa, (2021, Springer, Singapore).
[3] “FMOプログラムABINIT-MPの整備状況2021”, 望月祐志, 中野達也, 佐藤伸哉, 坂倉耕太, 渡邊啓正, 奥脇弘次, 大島聡史, 片桐孝洋, J. Comp. Chem. Jpn., 20 (2021) 132-136.
[4] “FMOプログラムABINIT-MPの整備状況2022”, 望月祐志, 中野達也, 坂倉耕太, 渡邊啓正, 佐藤伸哉, 奥脇弘次, 秋澤和輝, 土居英男, 大島聡史,片桐孝洋, J. Comp. Chem. Jpn., 21 (2022) 106-110.
Open Ver. 2 Rev. 8の高速化と大規模系対応
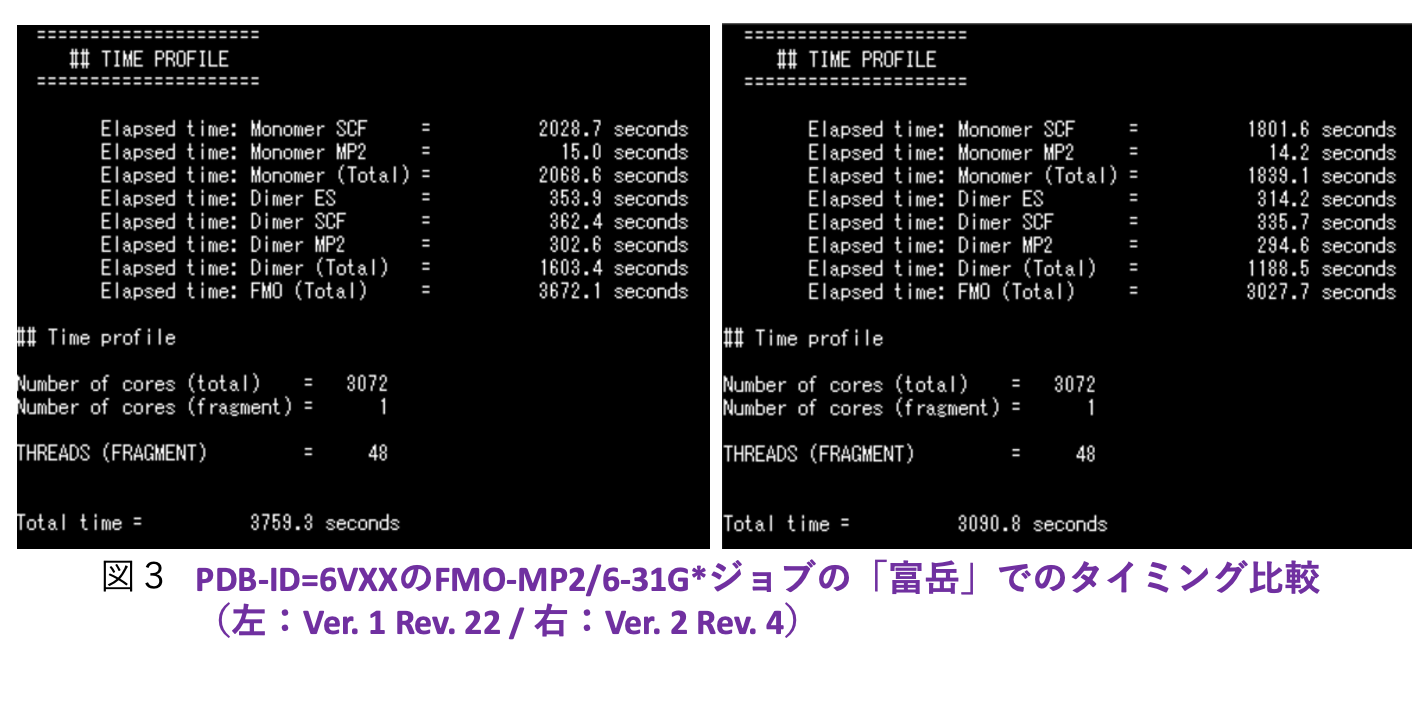
A64FX向けの高速化に関しては、「学際大規模情報基盤共同利用・共同研究拠点」(JHPCN)[5]の課題を通じてHPCの専門家とのコラボレーションの下で進めています。Rev. 4に対してRev. 8で追加的に実施した高速化作業は、レジスタスピルを減らすために2電子積分の生成ルーチン群にループ分割を行ったこと、Fock行列構築の最深部処理からif分岐を排除したことです。さらに、モノマーの自己無撞着場(SCC)の段階で2電子積分をバッファリングして各モノマーでのIn-core SCF計算を可としました。積分のバッファリングは、4つの軌道の角運動量の総和で指定できますが、実際の利用では対象系、基底関数の設定、ならびに使用する計算機のメモリの上限値に依存します。図1に、エイズウイルスのプロテアーゼにリガンド(ロピナビル)が結合した系のFMO-MP2/cc-pVDZジョブを名古屋大学の「不老」 Type I上でRev. 4とRev. 8で実行したタイミングを示します(MP2の積分変換は全段をDGEMMで処理)。Rev. 8での積分のバッファリングは、軌道角運動量の和で4までの積分が対象となっています。Rev. 4との比較では、モノマーSCFの加速が効いていることがわかりますが、ダイマーSCFも速くなっているのは積分ルーチンでのループ分割とFock行列計算の改良を反映しています。

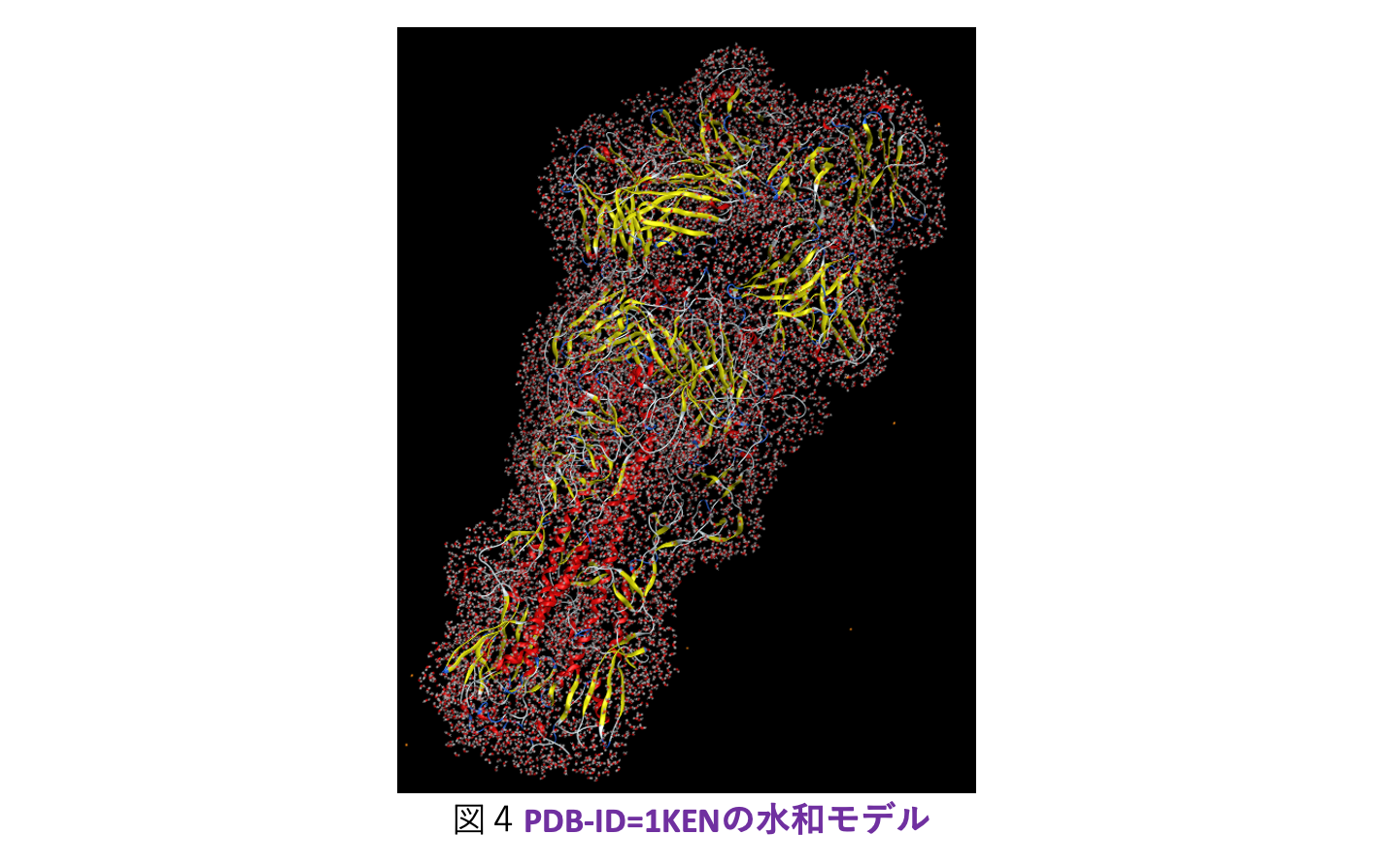
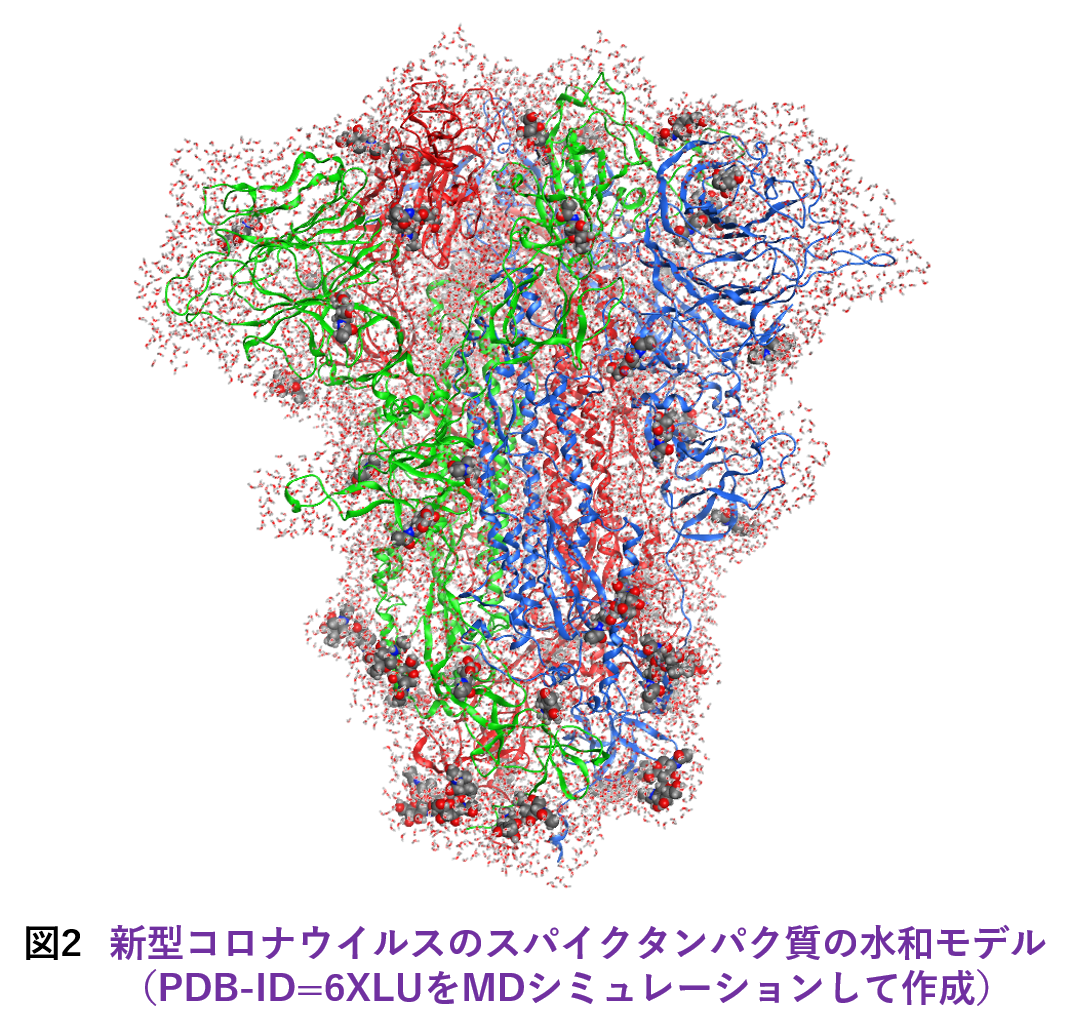
大規模系への対応についてですが、ターゲットとしては古典分子動力学(MD)シミュレーションによって生成された数千残基のタンパク質の水和モデルが主になると想定されます。そこで今回は発想を変え、ABINIT-MP本体の改造ではなく、フラグメント分割の前処理を行って実効的なフラグメント総数を減らすことを考えました。つまり、タンパク質に隣接する水分子は水和に伴う電荷移動や分極を考慮するために個別に扱うのに対し、遠方の水はクラスターとしてまとめるという形です。クラスタリングによってO原子が4,5個となるため、残基部分との粒度差が小さくなって並列処理のバランスの改善も期待できます。実際の処理はPythonスクリプトを使って行っていますが、新型コロナウイルスのスパイクタンパク質(3.3千残基)のような糖タンパク質の糖鎖部分も自動でフラグメント化できるようになっています。図2は、スパイクタンパク質(PDB-ID=6XLU)の水和モデルで、MD由来の構造サンプルのフラグメント総数は1.8万ですが、この前処理によって1万にまで低減できました。「富岳」の8ラック利用では、FMO-MP2/6-31G*ジョブが2.1時間で完走します。2020年度の段階では、Ver. 1 Rev. 22プログラムの制約からスパイクタンパク質の計算[6]は水和は無し(また糖も無し)での条件でやらざるを得ず、忸怩たる思いがありました。しかし、Ver. 2 Rev. 8ではフラグメント総数が2万程度となる数千残基のタンパク質の水和モデルのFMO-MP2計算は(「富岳」であれば)ルーチン的に実行可能になりました。なお、この前処理スクリプトのご利用については当面は応相談とさせていただきます。
[5] 「学際大規模情報基盤共同利用・共同研究拠点(JHPCN)」<https://jhpcn-kyoten.itc.u-tokyo.ac.jp/ja/>, 課題番号: jh210036-NAH, jh220010, jh230001.
[6] “Interaction analyses on SARS-CoV-2 spike protein based on fragment molecular orbital calculations”, K. Akisawa, R. Hatada, K. Okuwaki, Y. Mochizuki, K. Fukuzawa, Y. Komeiji, S. Tanaka, RSC Adv. 11 (2021) 3272-3279.
Open Ver. 2 Rev. 8の機能強化
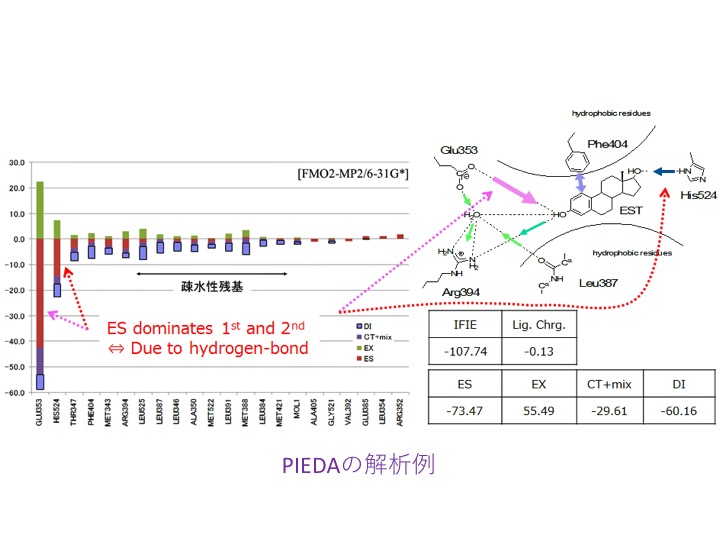
機能強化では、相互作用エネルギーの成分分解であるPIEDA [7,8]でMP2などの電子相関計算でのエネルギー低下を“DI”(Dispersion Interaction:分散相互作用)としている点に対する修正案として、LRD(Local Response Dispersion)[9]によって分散力系の安定化寄与を別に算定し、過剰なイオン性の低減などの相関補正の寄与と分離して評価可能としました。文献[10]では、LRD計算を別途行っていましたが、今回の改良で単一ジョブで計算可になりました[11]。構造活性相関(QSAR)や機械学習では、各寄与を分けて記述子とすることで予測精度が改善される可能性があります。また、環境静電ポテンシャル由来のRESP電荷を使った“ES項(Electrostatic Interaction:静電相互作用)”に対する参考値の算定機能オプションも入りました[11]。
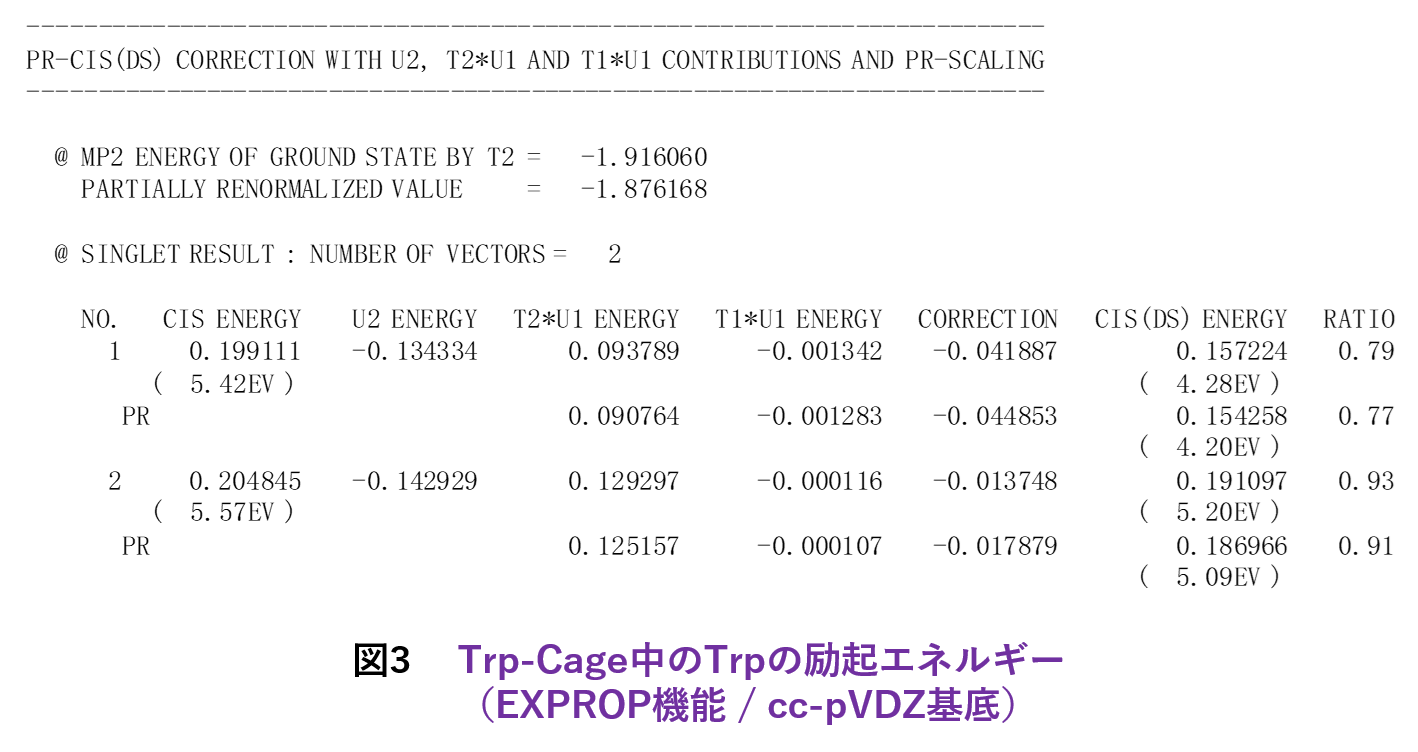
機能追加では、多層FMO [12]の文脈で1電子励起CI(CIS)[13]とMP2的なエネルギー補正(CIS(D):緩和エネルギーと差分相関エネルギーを評価)[14]が利用可能になりました[4]。&EXPROPネームリストで指定しますが、元々は立教大学の古いローカル版にあったモジュールを移植した機能になります。これにより、例えばTrp-Cageの興味領域であるTrp部分のみの励起エネルギーの算定ができます。CIS(D)では、MP2振幅を部分的に再規格化し、さらに相関導入による差分緩和エネルギーを考慮するオプション[15]、ならびに緩和エネルギーに準粒子・2次グリーン関数[16]の自己エネルギーシフトを入れるオプション[17]もあります。図3は、Trp-CageのTrpの励起エネルギーを文献[15]の機能で計算した例になります(基底関数はcc-pVDZ)。なお、CIS(D)の拡張でMP2振幅への自己エネルギーシフト[18]は、Rev. 8では未だサポートできていません。
2次グリーン関数の機能は単独でも使え、標的分子系の荷電状態には注意が要りますが、興味領域のイオン化エネルギーを計算できます(&EXPROPで指定)[4]。2次の自己エネルギーは、過大評価気味のクープマンスの値に比べると逆に過小評価に転じる傾向がありますが、pGW2スケーリング[19]を行うと妥当な値となります。なお、手動でのスケーリングパラメータも設定可能です。図4は、Trp-CageのTrpのイオン化エネルギーの算定値のリストです。

機能追加の最後は、分子凝集体でのフラグメント・ユニット間の励起状態のカップリングパラメータ[20,21]の算定です。この機能は、量子科学技術研究開発機構(QST)の藤田貴敏氏がローカル版のABINIT-MPに対して実装されていたモジュールの移植になり、&COUPLINGネームリストで設定します。光応答性分子の結晶状態での解析などに有用と思われます。
[7] “Pair Interaction Energy Decomposition Analysis”, D. G. Fedorov, K. Kitaura, J. Comp. Chem. 28 (2007) 222-237.
[8] “相互作用エネルギー成分分割解析機能PIEDAの実装とタンパク質-リガンド間の相互作用解析”, 塚本貴志, 加藤幸一郎, 加藤昭史, 中野達也, 望月祐志, 福澤薫, J. Comp. Chem. Jpn., 14 (2015) 1-9.
[9] “Density functional method including weak interactions: Dispersion coefficients based on the local response approximation”, T. Sato, H. Nakai, J. Chem. Phys. 131 (2009) 224104-1-12.
[10] “Fragment Molecular Orbital Based Interaction Analyses on Complexes Between SARS-CoV-2 RBD Variants and ACE2”, K. Akisawa, R. Hatada, K. Okuwaki, S. Kitahara, Y. Tachino, Y. Mochizuki, Y. Komeiji, S. Tanaka, Jpn. J. Appl. Phys. 60 (2021) 090901-1-5.
[11] “Enhancement of energy decomposition analysis in fragment molecular orbital calculations”, S. Matsuoka, K. Sakakura, Y. Akinaga, K. Akisawa, K. Okuwaki, H. Doi, Y. Mochizuki, to be submitted.
[12] “Multilayer Formulation of the Fragment Molecular Orbital Method (FMO)”, D. G. Fedorov, T. Ishida, K. Kitaura, J. Phys. Chem. A, 109 (2005) 2638-2646.
[13] “Configuration interaction singles method with multilayer fragment molecular orbital scheme”, Y. Mochizuki, S. Koikegami, S. Amari, K. Segawa, K. Kitaura, T. Nakano, Chem. Phys. Lett. 406 (2005) 283-288.
[14] “Parallelized integral-direct CIS(D) calculations with multilayer fragment molecular orbital scheme”, Y. Mochizuki, K. Tanaka, K. Yamashita, T. Ishikawa, T. Nakano, S. Amari, K. Segawa, T. Murase, H. Tokiwa, M. Sakurai, Theor. Chem. Acc. 117 (2007) 541-553.
[15] “Modification for spin-adapted version of configuration interaction singles with perturbative doubles”, Y. Mochizuki, K. Tanaka, Chem. Phys. Lett., 443 (2007) 389-397.
[16] “Propagators in Quantum Chemistry, Second Edition”, J. Linderberg, Y. Öhrn, (2004, Wiley, Hoboken NJ).
[17] “A practical use of self-energy shift for the description of orbital relaxation”, Y. Mochizuki, Chem. Phys. Lett., 453 (2008) 109-116.
[18] “Application of Dyson-corrected second-order perturbation theories”, Y. Mochizuki, Chem. Phys. Lett., 472 (2009) 143-148.
[19] “The parametrized second-order Green function times screened interaction (pGW2) approximation for calculation of outer valence ionization potentials”, C.-H. Hu, D. P. Chong, M. E. Casida, J. Elec. Spec. Rel. Phenom., 85 (1997) 39-46.
[20] “Molecular orbital calculation of biomolecules with fragment molecular orbitals”, S. Tsuneyuki, T. Kobori, K. Akagi, K. Sodeyama, K. Terakura, H. Fukuyama, Chem. Phys. Lett. 476 (2009) 104-108.
[21] “Charge-Transfer Matrix Elements by FMO-LCMO Approach: Hole Transfer in DNA with Parameter Tuned Range-Separated DFT”, H. Kitoh-Nishioka, K. Ando, Chem. Phys. Lett. 621 (2015) 96-101.
Open Ver. 2 Rev. 8 (2023年8月版)の開発者
Ver. 2 Rev. 8のABINIT-MP本体の整備・開発に関しては、中野達也氏(国立医薬品食品衛生研究所 医薬安全科学部)、坂倉耕太氏(計算科学振興財団)、望月祐志(立教大学 理学部:取り纏め役)の3名が主に関わりました。また、土居英男氏(立教大学 理学部)と奥脇弘次氏(JSOL&立教大学 理学部)は、入力データの整形ツールや「富岳」でのジョブ実行ツールなどPython系スクリプトを多数開発し、ABINIT-MPの周辺環境の整備に尽力しました。
NEC SX-Aurora TSUBASA向けのベクトル化
立教大学とNECとの共同研究によって、需要の多いFMO-MP2計算のSX-Aurora TSUBASA(SX-AT)向けのベクトル化チューニングが続けられてきました。2022年度までは佐藤伸哉氏(NECソリューションイノベータ)、2023年度からは加藤季広氏(NEC)が坂倉耕太氏(前出)と連携して実作業に当たっています。Ver. 1 Rev. 22では、ベクトル化により、新型コロナウイルスのメインプロテアーゼとN3リガンドの複合体PDB-ID=6LU7のFMO-MP2/6-31G*計算で改良前に比べて4.6倍の高速化が達成されています[3]。ベクトル化部分はVer. 2 Rev. 4にも移植されていますが、Rev. 8に対してはこれからになりますので、東北大学と大阪大学のSX-ATではVer. 2 Rev. 4での利用となります。なお、ベクトル実行ではDGEMM処理の加速が期待されるため、テンソル縮約のコストが支配的な3次摂動のFMO-MP3計算についても、チューニングを検討しています。
HPCI拠点での整備
HPCI拠点でのスパコンの更新が続いており、各拠点でサブシステムを複数持ち、CPUも富士通のA64FX、IntelのXeon、NECの SX-AT、AMD のEPYCとあり、さらにNVIDIAのGPUも搭載されることが増えているため、全ての環境に対応するのは次第に難しくなってきています。ただ、計算機能によってはflat MPIでしか並列化されていないものもありますので、できるだけOpenMP/MPIの混成並列のバイナリとMPI並列のバイナリの両方をサブシステムに応じて揃えるようにしたいと思っています。
文献[3]には、2021年度のHPCI拠点でのライブラリ整備状況が示されています。可視化解析のためにVer. 1 Rev. 22は今後も(できるだけ)残しますが、Ver. 2 Rev. 4を今回のVer. 2 Rev. 8と併存させるかどうかは拠点毎の事情・判断に依ります。なお、SX-ATについては2023年夏の時点ではVer. 2 Rev. 4までの対応です(上述)。
Ver. 2 Rev. 8のHPCI拠点のライブラリ整備予定は、「富岳」(R-CCS)、「不老」(名古屋大学)、「Wisteria」(東京大学)を2023年度内に済ませ、その後で他拠点へと進めていきます(スパコン更新の状況に依存)。
Open Ver.2系の今後の開発整備
Rev. 8の次は、2年後にリリースを目指すRev. 12を想定しています。開殻系の扱いは、現在のUHF/UMP2に加え、スピン汚染の問題のないROHF/ROMP2の準備を始めています。機能面での追加に関しては、相対論効果の考慮が必要な重原子の扱いに対し、これまでのMCP(Model Core Potential)[22]だけでなく、石村和也氏(X-ability)のSMASH [23]からECP(Effective Core Potential)モジュールを移植します。これにより、GAUSSIANプログラムで常用されるLANL2DZ [24-26]が使えるようになりますので、例えばシスプラチン結合DNAの扱いでQM/MMモデルとの直接比較などが可能となり、利用者の利便性が向上するはずです。
Rev. 12での高速化については、GPU対応との関係から垂直漸化式関係[27]に基づいて軌道タイプの組み合わせに応じて自動生成された2電子積分ルーチン群の再構成を行う予定です。GPU化については、FMO機能を省いたHF/SCF計算のミニアプリを成瀬彰氏(NVIDIA)がチューニングされ、Xeon単独に比してGPUによって6-8倍の加速が得られた経験があります[28]。このチューニングでは、NVIDIA-GPUのCUDAコアを埋めるために積分ルーチン群が軌道角運動量に応じてまとめられ、性能向上に繋がりました。2023年夏時点で、この方針をベースにABINIT-MP本体の改造をどう進めていくか検討しているところです。
大学のHPCI拠点では、いくつかの異なる構成のサブシステムが設置されることが多くなってきていますので、GPU化とは別にA64FX、SX-ATあるいはXeon系の高速化も引き続き取り組んでいくことになりますが、上記の積分ルーチンの再構成は手動・自動チューニング作業の能率を上げると期待しています。
大規模系対応については、Ver. 2 Rev. 8では見送った作業配列群の整理を再度行う予定です。これにより、水和タンパク質モデルで元のフラグメント数が4万程度の系でも扱えるようにしたいと考えています(水のクラスタリング処理は併用)。
[22] “Fragment molecular orbital calculations on large scale systems containing heavy metal atom”, T. Ishikawa, Y. Mochizuki, T. Nakano, S. Amari, H. Mori, H. Honda, T. Fujita, H. Tokiwa, S. Tanaka, Y. Komeiji, K. Fukuzawa, K. Tanaka, E. Miyoshi, Chem. Phys. Lett., 427 (2006) 159-165.
[23] https://sourceforge.net/projects/smash-qc/
[24] “Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg”, P. J. Hay, W. R. Watt, J. Chem. Phys. 82 (1985) 270-283.
[25] “Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi”, W. R. Watt, P. J. Hay, J. Chem. Phys. 82 (1985) 284-298.
[26] “Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals”, P. J. Hay, W. R. Watt, J. Chem. Phys. 82 (1985) 299-310.
[27] “Efficient recursive computation of molecular integrals over Cartesian Gaussian functions”, S. Obara, A. Saika, J. Chem. Phys., 84 (1986) 3963-3974.
[28] “HF計算に特化したABINIT-MPミニアプリのGPU化の試み”, 望月祐志, 坂倉耕太, 成瀬彰, 古家真之介, 下川辺隆史, 芝隼人, 大島聡史, 星野哲也, 片桐孝洋, 理論化学討論会2023, 2023/5/19 (横浜).
謝辞
ABINIT-MP Open Ver. 2系のA64FX向けの高速化と大規模系への対応は、JHPCN [5]の継続課題{jh210036-NAH, jh220010, jh230001}「FMOプログラムABINIT-MPの高速化と超大規模系 への対応」(代表者:望月祐志)の中で展開されています。この中で、片桐孝洋先生(名古屋大学 情報基盤センター)、大島聡史先生(九州大学 情報基盤研究開発センター)にはHPCの専門的なアドバイスをいただいています[3,4]。2023年度からは、星野哲也先生(名古屋大学 情報基盤センター)と滝沢寛之先生(東北大学 サイバーサイエンスセンター)にもご支援いただいています。対象とするスパコンとしては、2021-2022年度は名古屋大学の「不老」 Type Iを作業拠点としたA64FX向けの対応のみでしたが、2023年度からはNECと立教大学とのこれまでの共同研究をベースに、東北大学の「AOBA-A/S」をプラットフォームとしたSX-AT向けの活動(滝沢先生関係)も公式に行っています。
「富岳」での大規模なテスト計算は、感染症対策関係のHPCI課題{hp210026, hp220025, hp220352, hp230017}(代表者:望月祐志)の中で行っています。また、小規模FMOジョブを大量に実行して粗視化シミュレーションの一種である散逸粒子動力学(DPD)のパラメータを決めるアプローチ[29,30]を「富岳」上で推進する課題{hp210261, hp230016}(代表者:望月祐志)とも連動しています。資金面では、2022年度から立教SFRの支援を受けています。
SX-AT向けのベクトル化による高速化チューニング、ならびに解析機能の向上については長年NEC様からご支援をいただいています。また、複数の企業様からの立教大学への指定寄付を得て、ABINIT-MPプログラムの研究開発と維持ができています。
励起状態でのカップリングの算定機能では、モジュールを提供くださった藤田貴敏氏(前出)、ならびに機能の必要性をご議論いただき、さらに移植にご支援いただいた小沢拓氏と新田浩也氏(共にJSOL)にも感謝したいと思います。
HPCI拠点へのABINIT-MPのライブラリ整備やハンズオンセミナーでは、(一財)高度情報科学技術研究機構(RIST)に継続的にお世話になっています。
最後に、「計算工学ナビ」サイトでのABINIT-MP関係の情報公開で長年ご支援いただいている東京大学生産技術研究所の加藤千幸先生、同革新的シミュレーション研究センターにも謝意を表します。
[29] “Fragment Molecular Orbital-based Parameterization Procedure for Mesoscopic Structure Prediction of Polymeric Materials”, K. Okuwaki, Y. Mochizuki, H. Doi, T. Ozawa, J. Phys. Chem. B, 122 (2018) 338-347.
[30] “フラグメント分子軌道(FMO)法を用いた散逸粒子動力学シミュレーションのための有効相互作用パラメータ算出の自動化フレームワーク”, 奥脇弘次, 土居英男, 望月祐志, J. Comp. Chem. Jpn., 17 (2018) 102-109.
コンタクト
ABINIT-MPのOpenシリーズのご利用、あるいはプログラム開発にご相談のある方は、取り纏め責任者の立教大学の望月祐志(fullmoon -at- rikkyo.ac.jp)にメールにてご連絡いただければ適宜対応させていただきます(-at-を@に変換してください)。