ABINIT-MP Openシリーズ (Ver.2 Rev.12)
※2023年8月版(Ver.2 Rev.8)に関するページはこちらです
※2020年6月版(Ver.1 Rev.22)に対応するBSV(BioStation Viewer)のWindows用インストーラはこちらからダウンロードできます
はじめに
ABINIT-MP [1,2]のOpenシリーズのVer. 2系は、2021年9月のRev. 4 [3]、2023年8月のRev. 8 [4,5]に次いで、今回(2026年3月)が3回目のRev. 12 [6,7]となります。Ver. 2系は、2021年度からJHPCN [8]のABINIT-MP関係の課題(代表:望月祐志)の中で計算機科学の高性能計算(HPC)の専門家グループとのコラボレーションの下、特に高速化が進められてきました(速度向上については、Ver. 2 Rev. 8のページを参照)。実は、Rev. 12はRev. 8から高速化に関しては、NECのSX Aurora-TSUBASA(SX-AT)向けベクトル化チューニング以外の進捗はありません。その代わり、タンパク質の液滴モデルへの対応力を大幅に高めています。また、水のクラスターですが4万フラグメントの扱いも可能となりました。機能面でも幾つかの改良が入っており、ネームリストも更新されていますので、旧入力データajfを転換するPythonスクリプトも併せて提供します。
[1] “Electron-correlated fragment-molecular-orbital calculations for biomolecular and nano systems”, S. Tanaka, Y. Mochizuki, Y. Komeiji, Y. Okiyama, K. Fukuzawa, Phys. Chem. Chem. Phys. 16 (2014) 10310-10344.
[2] “The ABINIT-MP Program”, Y. Mochizuki, T. Nakano, K. Sakakura, Y. Okiyama, H. Watanabe, K. Kato, Y. Akinaga, S. Sato, J. Yamamoto, K. Yamashita, T. Murase, T. Ishikawa, Y. Komeiji, Y. Kato, N. Watanabe, T. Tsukamoto, H. Mori, K. Okuwaki, S. Tanaka, A. Kato, C. Watanabe, K. Fukuzawa (pp.53-67) in “Recent Advances of the Fragment Molecular Orbital Method – Enhanced Performance and Applicability”, ed. by Y. Mochizuki, S. Tanaka, K. Fukuzawa, (2021, Springer, Singapore).
[3] “FMOプログラムABINIT-MPの整備状況2021”, 望月祐志, 中野達也, 佐藤伸哉, 坂倉耕太, 渡邊啓正, 奥脇弘次, 大島聡史, 片桐孝洋, J. Comp. Chem. Jpn., 20 (2021) 132-136.
[4] “FMOプログラムABINIT-MPの整備状況2022”, 望月祐志, 中野達也, 坂倉耕太, 渡邊啓正, 佐藤伸哉, 奥脇弘次, 秋澤和輝, 土居英男, 大島聡史,片桐孝洋, J. Comp. Chem. Jpn., 21 (2022) 106-110.
[5] “FMOプログラムABINIT-MPの整備状況2023”, 望月祐志, 中野達也, 坂倉耕太, 奥脇弘次, 土居英男, 加藤季広, 滝沢寛之, 成瀬彰, 大島聡史, 星野哲也, 片桐孝洋, J. Comp. Chem. Jpn., 23 (2024) 4-8.
[6] “ABINIT-MPプログラムの現状と今後”, 望月祐志, 中野達也, 坂倉耕太, 土居英男, 奥脇弘次, 加藤季広, 滝沢寛之, 大島聡史, 星野哲也, 片桐孝洋, J. Comp. Chem. Jpn., 23 (2024) 85-97.
[7] “FMOプログラムABINIT-MPの整備状況2025”, 望月祐志, 中野達也, 坂倉耕太, 土居英男, 奥脇弘次, 福澤薫, 加藤季広, 成瀬彰, 星野哲也, 大島聡史, 滝沢寛之, 中島研吾, 片桐孝洋, J. Comp. Chem. Jpn., 25 (2026) 7-14.
[8] 「学際大規模情報基盤共同利用・共同研究拠点(JHPCN)」, 課題番号: jh210036-NAH, jh220010, jh230001, jh240001, jh250002
Open Ver. 2 Rev. 12の特徴
Rev. 8に対するRev. 12の更新は,超大規模系の対応力強化と機能の追加が主なところです[7]。以下、説明します。
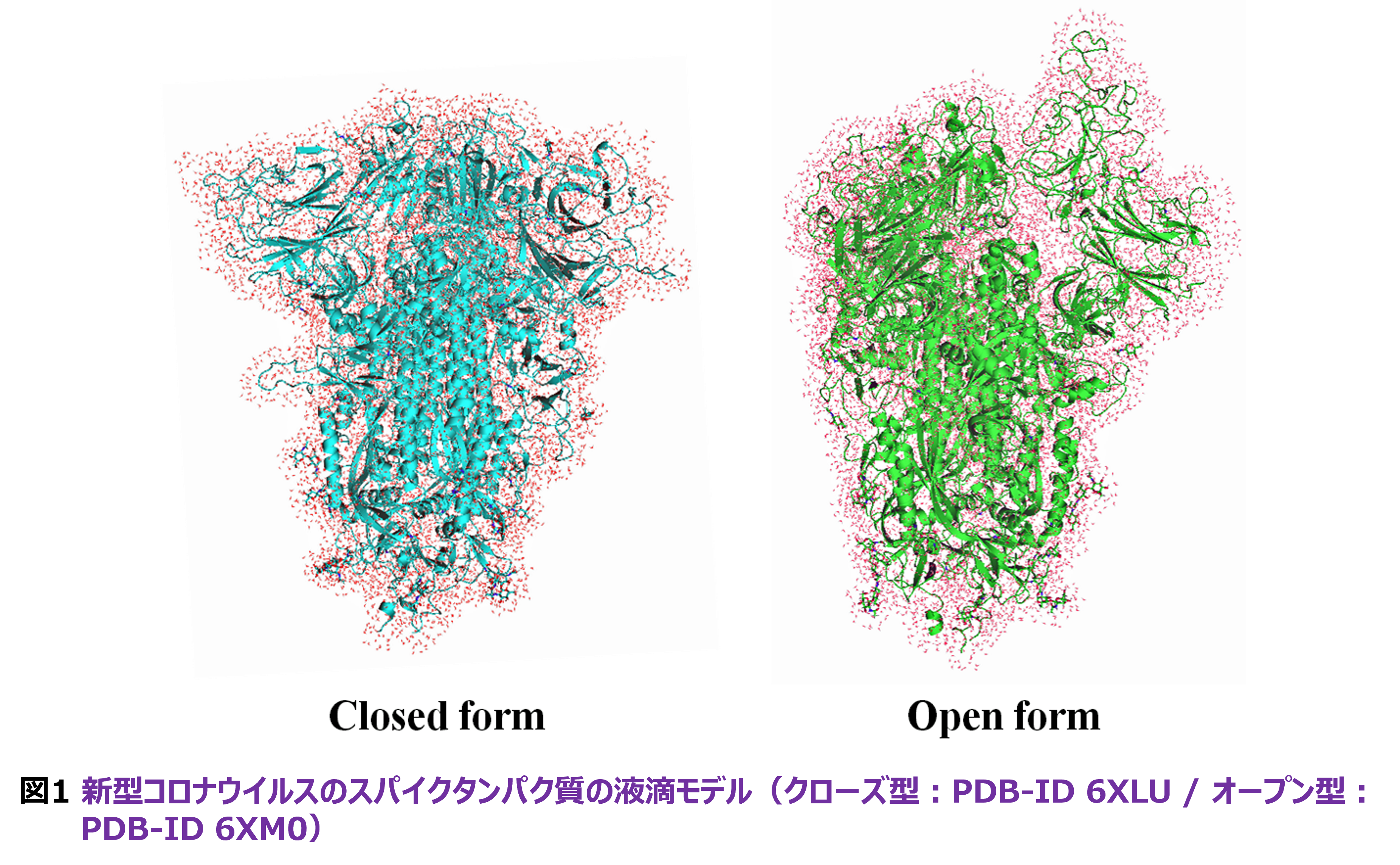
先ず、超大規模系についてです。「富岳」の時代になり、分子動力学(MD)シミュレーションの軌跡から切り出される液滴モデルを一括してFMO計算し、相互作用解析を統計的に行えるにようになっています(「富岳」での最初の事例は[8])。いわゆる、MD-FMO連携計算のアプローチです。この液滴モデルでは、中性化のためのイオンを温存することが通常ですが、交換反発が考慮されない古典力場の「癖」により、イオンと残基の側鎖との距離が近くなり過ぎることがあります。これにより、モノマーやダイマーの段階でのハートリーフォック(HF)の収束不可が発生してしまい、FMOジョブの完走が得られません。そこで、液滴モデルのPDBデータをスキャンして、イオンを抜き出して点電荷で近似する仕組みを導入しました。処理はPythonスクリプトで行います。実証例としては、Rev. 8のページでも紹介しています新型コロナウイルスのスパイクタンパク質の液滴モデルに適用しました。図1は、受容体結合領域(RBD)が閉じたクローズ型(PDB-ID 6XLU)と開いたオープン型(同6XM0)を示していますが、特にオープン型で未完走が多発しました。乱暴にイオンを全て除去すると完走率が上がりますが、タンパク質内の荷電バランスが変わるために残基間の相互作用エネルギーも影響を受けてしまいます。文献[9]では、こうした結果を示すと共に、液滴モデル1つあたり「富岳」の8ラック(3,072ノード)で僅か2時間でFMO-MP2/6-31G(d)ジョブが処理できることを報告しています。MD-FMOの液滴モデルでの点電荷近似は、立教大学での応用計算では2024年度から常用しており、相互作用エネルギーの統計的算定を安定して行えるようになっています。
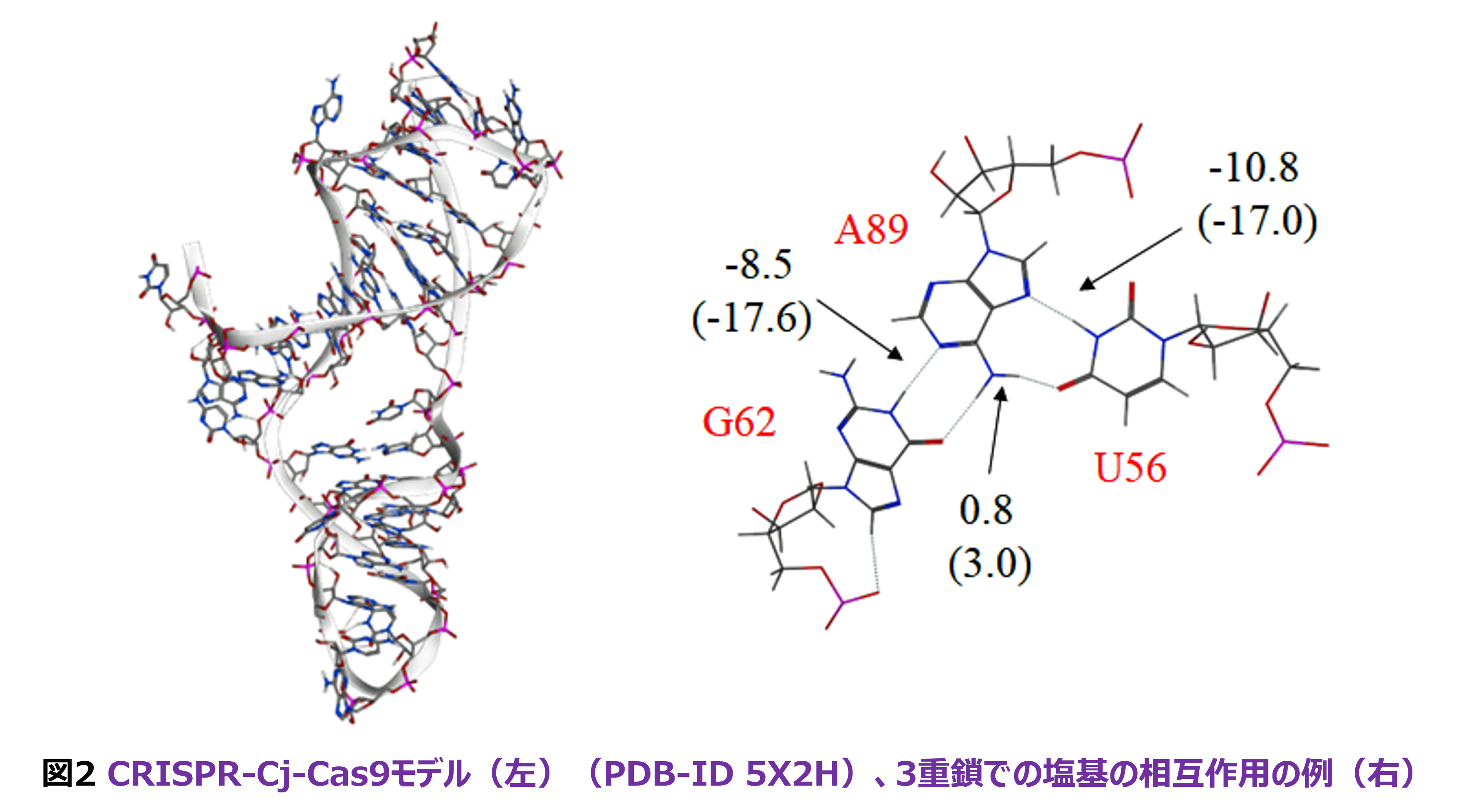
Pythonスクリプトに関してはフラグメント分割でも改良が進んでいます。特に、タンパク質にDNAやRNAが共存する系の扱いは、これまでは手動分割が必要でしたが、これが自動化されました。実テストの系では、タンパク質/DNA/RNAが複合するCRISPR系の最小モデルであるCj-Cas9(PDB-ID 5X2H)を選びました。図2左に構造を示しますが、上部にある塩基ユニットが3重鎖を形成する部分が注目領域の1つになります。図2右は、その一例でWatson-Crick型ではなくHoogsteen型の水素結合がA-U間にあることがわかります。水素結合のエネルギーは、FMO-MP2/cc-pVDZで算定したサンプルの平均値ですが、PIEDAの拡張機能[10]を用いて静電項(ES)をFMO-RESP電荷を使った静電エネルギー表式の値で置き換えた値を括弧無しで示しています。文献[10]の中では、スタック塩基対モデルで過大評価の是正を明示していますが、このCRISPR-Cas9の系でも有効であると考えています。
ここで、地道なメモリ削減によって超大規模計算が可能になったことを記しておきます。図3に示す立方体状のモデルクラスターですが、40,587個の水分子のFMO-MP2/6-31G(d)ジョブが、「富岳」と同じA64FXの「不老」 Type I(2026年3月に退役)の半ラック(192ノード)の計算資源を使って10時間で完走しました。フラグメントあたり、6スレッド(OpenMP)を立て、1152プロセス(MPI)の混成での実行でした。この事実は、ABINIT-MPが(素で)数万フラグメントのFMO計算を処理できる段階に至ったことを示しています。

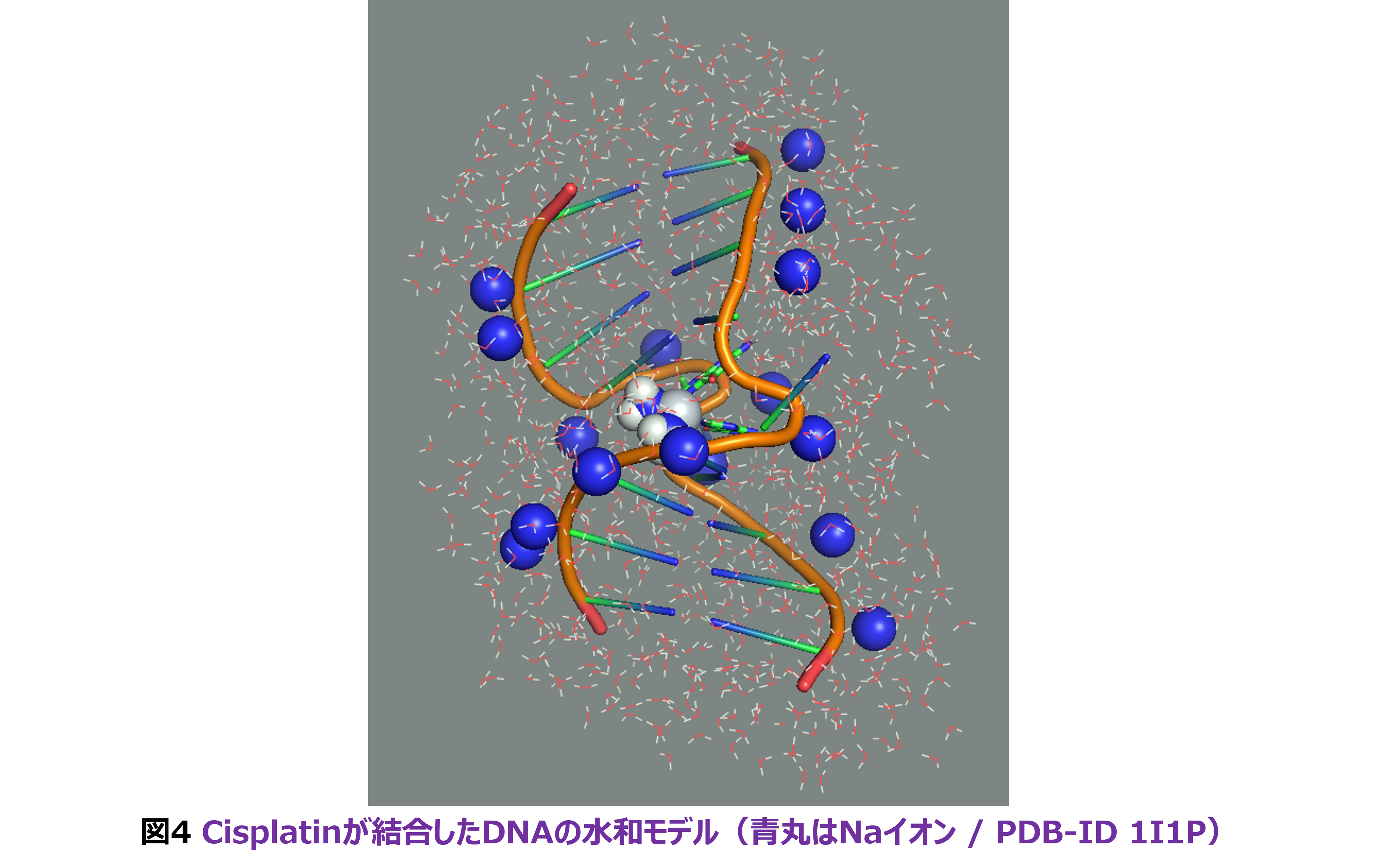
機能強化に話を移します。1つは、これまでのスカラー相対論的なモデル内殻ポテンシャル(MCP)[11]に加え、石村和也氏のSMASH [12]から有効内殻ポテンシャル(ECP)を移植して利用可能としたことです。これにより、ネームリストは&MCPから&RELPOTに変わりました。ポテンシャルとしては、LANL2DZ [13-15]をデフォルト想定していますが、内蔵セットだけでなくBasis Set Exchangeサイト[16]からダウンロードしたセット(GAMESS-USフォーマット)も読み込み可能です。ただ、hポテンシャルには未だ対応していないためアクチノイドの計算には制限があります。ECPのテストとして、文献[11]で扱った小型DNA片にCisplatinが結合した構造(PDB-ID 1I1P)の水和モデル(図4)を計算し、塩基ユニット間で妥当な相互作用エネルギーが得られることを確認しています。
機能強化の2つめは多層FMOの扱いで、Rev. 8でのCIS/CIS(D)による励起エネルギー算定[17,18]についてです。差分相関エネルギーに対するMP2振幅の部分再規格化と相関導入に依る差分緩和エネルギーの補正[19]、軌道緩和エネルギーに対する自己エネルギー修飾[20]に加え、MP2振幅にも自己エネルギー修飾[21]を追加したことです。CIS系の「対角化→摂動」の範囲内ですが、若干の近接縮退を伴う励起状態のエネルギー算定の信頼性が向上しています。
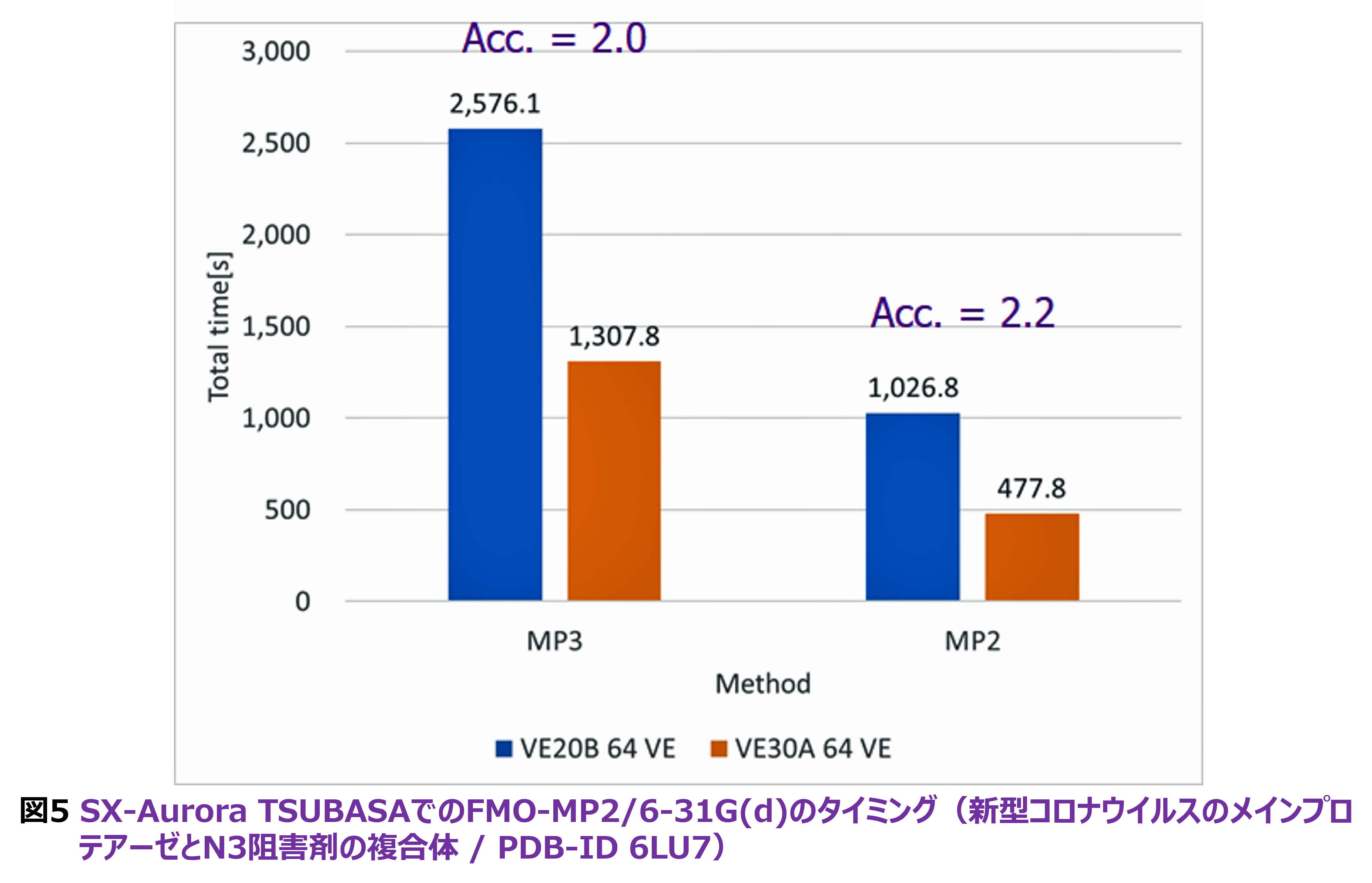
速度向上に関しては、SX-ATでのMP3計算[22]の改良があります。MP3計算では、”4粒子-2空孔”の寄与の処理の最深部でDAXPYを用いますが、これをインライン展開としました。SX-ATのVE20BとVE30Aの新旧のベクトルエンジンを比較で用い、新型コロナウイルスのメインプロテアーゼとN3リガンドの複合体(PDB-ID 6LU7)の6-31G(d)基底で測定した結果を図5に示します。VE30Aはコア数16とVE20Bの8の2倍となり、さらにL3キャッシュが追加されていますが、FMO-MP2での加速は2.2倍となって両者の相乗効果が見えます。FMO-MP3でも2.0倍の加速が得られていますが、インライン展開前のコードでは1.6倍に留まってしまいます。相対コストはVE30Aで2.7倍ですので、SX-AT上でのMP3計算の実用性が高まりました。
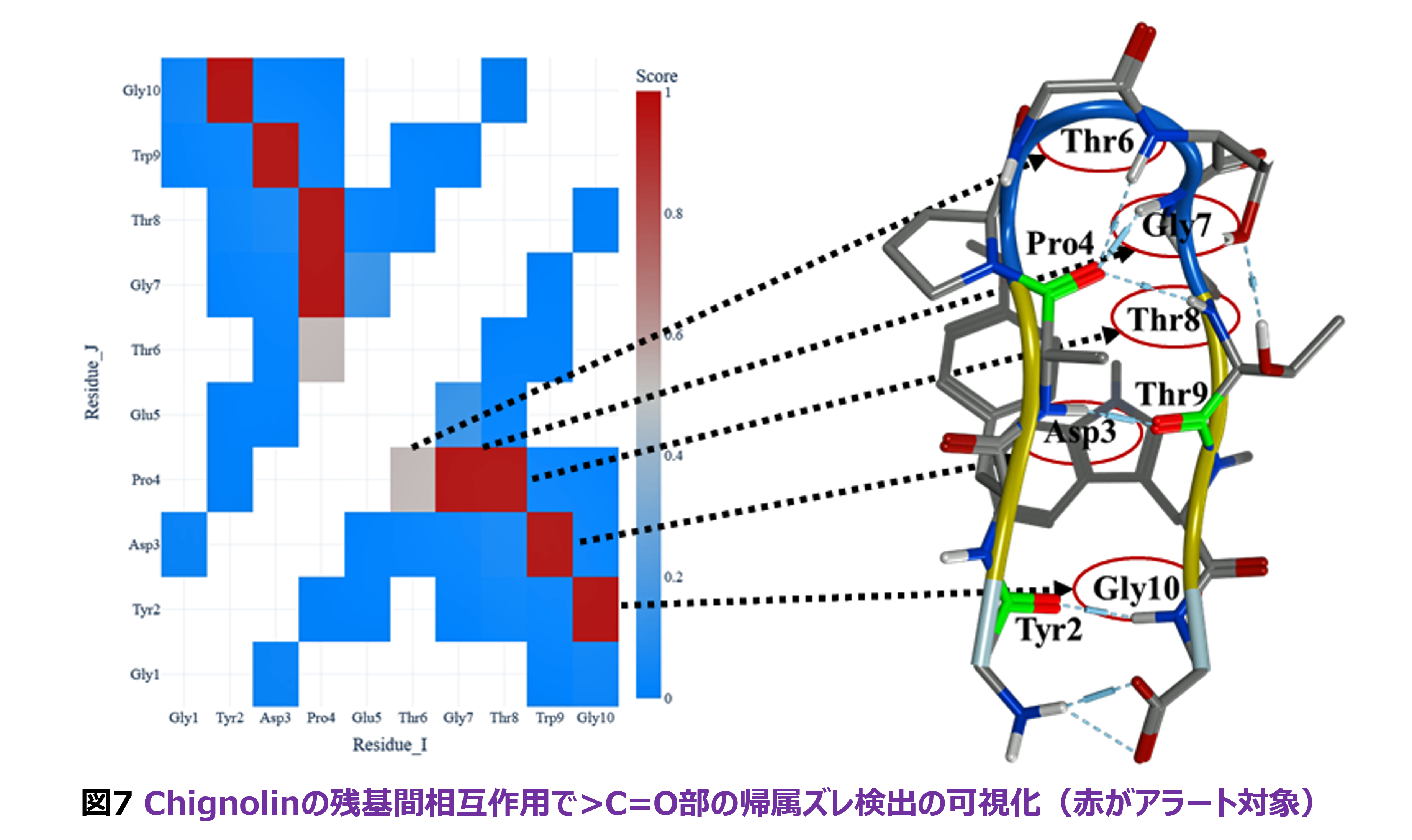
MD-FMO連携計算では、入力データの作成~ジョブの実行~結果データの解析で様々なPythonスクリプトが使われており、液滴モデルの点電荷近似とフラグメント分割については上記していますが、その他のスクリプトについても触れておきます。1つめは、いわゆるサロゲートモデリング用のスクリプト(仮名称はSSIFIEE)[23]です。これは、6-31G(d)やcc-pVDZ基底での相互作用エネルギーをsp関数のみの3-21G基底での結果を機械学習で「補完」して定量的に予測しようというものです。図6に、Chignolin(PDB-ID 1UAO)での6-31G(d)の結果を予測した決定係数(R2)をヒートマップ的に示します。多くの残基対に関して十分な精度が出ていることがわかります。ただ、cc-pVDZについては改良の余地があります。2つめは、フラグメント分割がペプチド結合ではなく、側鎖を持つCαとカルボニル炭素の間で行われることに伴って生じ得る水素結合に関わる残基の「帰属ずれ」の自動検出ツール(名称はSARASA)[24]です。図7は、Chignolinでのテストの結果で、赤く図示されている残基対は「帰属ずれ」がアラートされています。この「ずれ」は、FMOの初心者ではしばしば混乱を招くだけでなく、自動データ解析の際にも問題となりますが、SARASAを使えば回避できます。現在、配位結合に対する「帰属ずれ」にも対応しつつあります。

最後にお断りになりますが、これまで同様、入力データのajfのコンバータを除くPythonツール群は(諸事情から)オンデマンドでの提供とさせていただきます。
[8] “Statistical interaction analyses between SARS-CoV-2 main protease and inhibitor N3 by combining of molecular dynamics simulation and fragment molecular orbital calculation”, R. Hatada, K. Okuwaki, K. Akisawa, Y. Mochizuki, Y. Handa, K. Fukuzawa, Y. Komeiji, Y. Okiyama, and S. Tanaka, Appl. Phys. Express 14 (2021) 027003-1-5.
[9] “Large-scale FMO-MP2 calculations of the spike protein droplet model”, H. Doi, T. Nakano, K. Sakakura, K. Akisawa, K. Okuwaki, Y. Hirano, E. Yamamoto, K. Yasuoka, S. Ohshima, T. Katagiri, and Y. Mochizuki, J. Comp. Chem., 46 (2025) e70052-1-6.
[10] “Enhancement of energy decomposition analysis in fragment molecular orbital calculations”, S. Matsuoka, K. Sakakura, Y. Akinaga, K. Akisawa, K. Okuwaki, H. Doi, Y. Mochizuki, J. Comp. Chem., 45 (2024) 898-902.
[11] “Fragment molecular orbital calculations on large scale systems containing heavy metal atom”, T. Ishikawa, Y. Mochizuki, T. Nakano, S. Amari, H. Mori, H. Honda, T. Fujita, H. Tokiwa, S. Tanaka, Y. Komeiji, K. Fukuzawa, K. Tanaka, E. Miyoshi, Chem. Phys. Lett., 427 (2006) 159-165.
[12] https://sourceforge.net/projects/smash-qc/
[13] “Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg”, P. J. Hay, W. R. Watt, J. Chem. Phys. 82 (1985) 270-283.
[14] “Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi”, W. R. Watt, P. J. Hay, J. Chem. Phys. 82 (1985) 284-298.
[15] “Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals”, P. J. Hay, W. R. Watt, J. Chem. Phys. 82 (1985) 299-310.
[16] https://www.basissetexchange.org/
[17] “Configuration interaction singles method with multilayer fragment molecular orbital scheme”, Y. Mochizuki, S. Koikegami, S. Amari, K. Segawa, K. Kitaura, T. Nakano, Chem. Phys. Lett. 406 (2005) 283-288.
[18] “Parallelized integral-direct CIS(D) calculations with multilayer fragment molecular orbital scheme”, Y. Mochizuki, K. Tanaka, K. Yamashita, T. Ishikawa, T. Nakano, S. Amari, K. Segawa, T. Murase, H. Tokiwa, M. Sakurai, Theor. Chem. Acc. 117 (2007) 541-553.
[19] “Modification for spin-adapted version of configuration interaction singles with perturbative doubles”, Y. Mochizuki, K. Tanaka, Chem. Phys. Lett., 443 (2007) 389-397.
[20] “A practical use of self-energy shift for the description of orbital relaxation”, Y. Mochizuki, Chem. Phys. Lett., 453 (2008) 109-116.
[21] “Application of Dyson-corrected second-order perturbation theories”, Y. Mochizuki, Chem. Phys. Lett., 472 (2009) 143-148.
[22] “Large-scale FMO-MP3 calculations on the surface proteins of influenza virus, hemagglutinin (HA) and neuraminidase (NA)”, Y. Mochizuki, K. Yamashita, K. Fukuzawa, K. Takematsu, H. Watanabe, N. Taguchi, Y. Okiyama, M. Tsuboi, T. Nakano, S. Tanaka, Chem. Phys. Lett., 493 (2010) 346-352.
[23] “Prediction of quantitative interaction energy from low-cost FMO calculation by machine learning”, H. Doi, R. Yoshine, S. Matsuoka, K. Okuwaki, Y. Mochizuki, Jpn. J. Appl. Phys., 64 (2025) 077001-1-5.
[24] “Automatic detection of interacting carbonyl oxygens in peptide bond for the fragment molecular orbital method”, R. Yoshine, H. Doi, K. Okuwaki, Y. Mochizuki, Chem. Lett., 55 (2026) upaf238-1-4.
Ver. 1系FMODD版とHPCI拠点での整備
2024年秋からFMODDグループ[25]との交流が再開し、Ver. 1 Rev. 22系[26]のFMODD独自改良版とVer. 2系との「将来的な再統合」を意識した連携活動が行われるようになっています。最初の実績としては、2025年3月に凍結領域(FD)近似下での部分構造最適化[27]が可能な2024年度版(V1DD-24)を大阪大学、理研R-CCSにVer. 2系と併存の形でライブラリ登録しています。また、リリース予定のFMODDの2026年度版には文献[10]の拡張PIEDAが移植されている一方、本Ver. 2 Rev. 12にFMO-HFエネルギー微分のFMODD版の改良が反映されています。
文献[7]に明記していますが、ABINIT-MPは北海道大学~九州大学までの国内HPCI拠点(スーパーコンピュータセンター)ではライブラリプログラムとして利用できるようになっており、各所での機種の更新に対しても可能な限り対応しています。Ver. 2 Rev. 12についても、「富岳」を皮切りに2026年度にライブラリ整備を行います。
[25] https://fmodd.jp/
[26] “FMOプログラムABINIT-MPの整備状況2020”, 望月祐志, 坂倉耕太, 渡邊啓正, 奥脇弘次, 加藤幸一郎, 渡辺尚貴, 沖山佳生, 福澤薫, 中野達也, J. Comp. Chem. Jpn., 19 (2020) 142-145.
[27] “Geometry Optimization using the Frozen Domain and Partial Dimer Approach with the Fragment Molecular Orbital Method: Implementation, Benchmark, and Application for Ligand-Binding Site of Proteins”, K. Okuwaki, N. Watanabe, K. Kato, C. Watanabe, N. Nakayama, A. Kato, Y. Mochizuki, T. Nakano, T. Honma, and K. Fukuzawa, J. Chem. Inform. Model., 64 (2024) 9449-9458.
Open Ver. 2 Rev. 12 (2026年3月版)の主開発者
Ver. 2 Rev. 12のABINIT-MP本体の整備・開発では、中野達也氏(高度情報科学技術研究機構 利用支援部)、坂倉耕太氏(大阪大学 D3センター)、望月祐志(立教大学 理学部:取り纏め役:本稿執筆者)の3名が主に関わっています。また、土居英男氏(立教大学 理学部)と奥脇弘次氏((株)JSOL&立教大学 理学部&大阪大学 大学院薬学研究科)は、Python系スクリプト群を開発しています。
今後の開発・整備
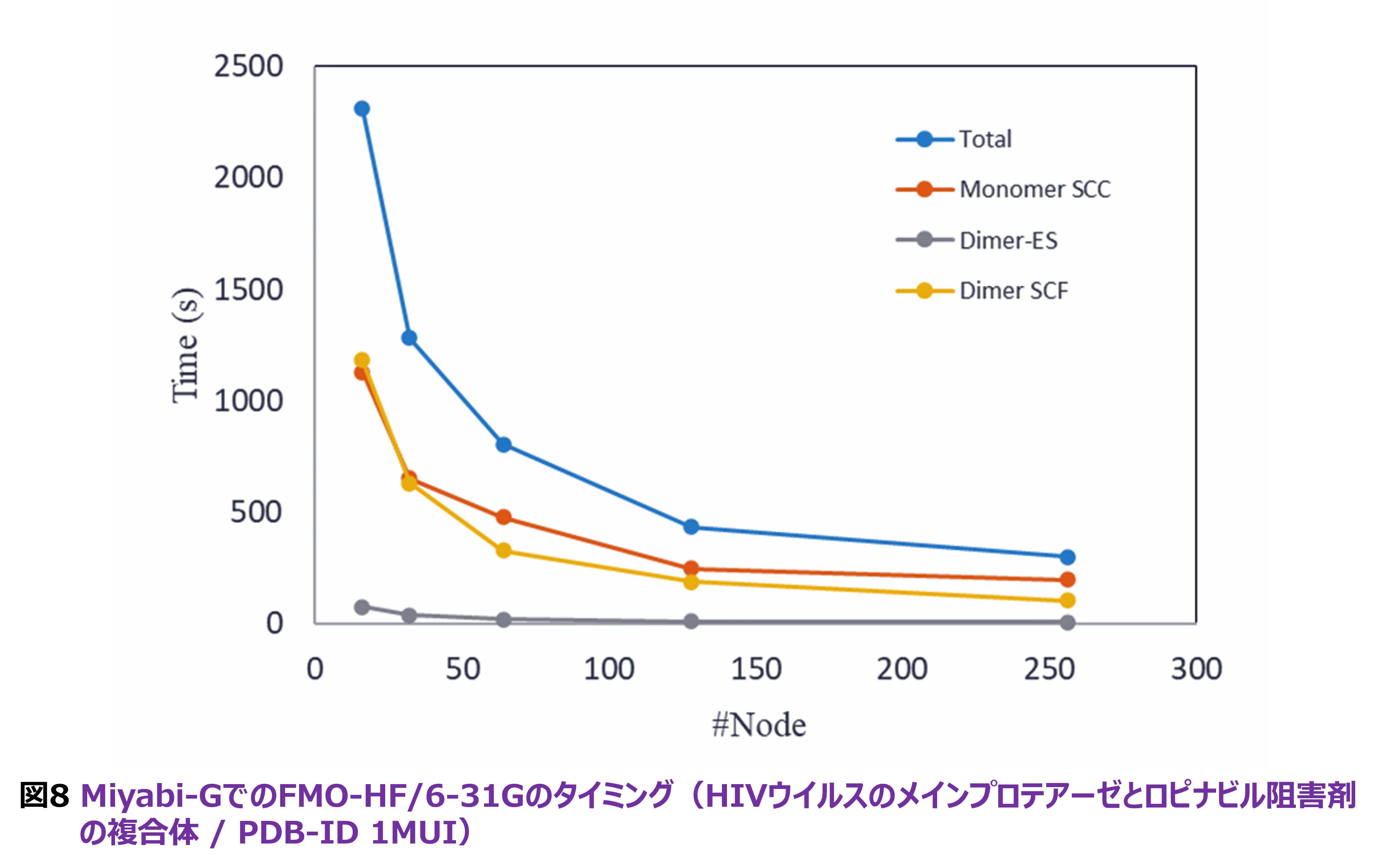
HPCI拠点では、近年GPU搭載のサブシステムが増えてきています。また、2030年に稼働が予定されている「富岳NEXT」では、1ノードに2基のCPU(富士通MONAKA-X)と4基のGPU(NVIDIA製は確定)が搭載されます。このため、ABINIT-MPでもGPU対応を急いでいます[5-7]。2026年春時点では、小原の垂直漸化式関係(VRR)[28]に基づく2電子積分周りの再構成とOpenACCディレクティブの挿入による改造がFMO-HFで(ほぼ)完了し、MP2エネルギーの積分変換は新規コードによる試行が進んでいるところです。FMO-HFに関しては、NVIDIAのGH200をノードに1基積むJCAHPCのMiyabi-Gでの多ノードテストが行われています[29]。図8は、HIVウイルスのプロテアーゼと阻害剤のリトナビルの複合体(PDB-ID 1MUI)の6-31G基底でのFMO-HFジョブのタイミングを16ノード起点で256ノードまで示したものですが、スケーリングとしては及第レベルです。MP2を新コードとしているのは、既存のDGEMMベースのコードではcuBLASを使っても好ましい加速が得られなかったためです。MPS/MIGなどのGPUのテンソルコア資源の実行時の分割も試みながら性能評価をしていますが、CPUのみの実行に比較してGPUも使うとFMO-MP2/6-31Gジョブで5倍程の加速が見えている段階です。チューニングが一段落した段階で、Ver. 2 Rev. 12の更新としてFMO-MP2エネルギー計算までをGPU対応としてRev. 16を2026年度内にリリースする予定で、Miyabi-G、名古屋大学の「不老・弐」タイプII、九州大学の「玄界」ノードグループB等でライブラリ公開します。なお、Ver. 2系はRev. 16で終了します。
文献[7]中に記していますが、GPU対応は米国のM. Gordonグループが大きく先行しており、GAMESS-USだけでなくEXESSなるGPU特化型の新しいプログラムも開発していますが、後者はオーストラリアのG. M. J. Barca氏らが主に進めています(文献情報は[7]のリンクを参照)。EXESSの本質的な特徴は、2電子積分を恒等分解(RI)近似で扱っていることで、これによって行列積ベースの処理が基本となるためGPUでの高速実行が可能となっています。ABINIT-MPにも2電子積分を近似するコレスキー分解(CD)が実装[30]されていますが、RIが最適化された基底での展開となるのに対し、CDでは自基底の積が基本となるために演算オーダーが1つ上がってしまい、加速と精度のバランス取りが困難であり、近年ではほとんど使われなくなりました。そこで、RIの実装で先行するPAICS [31,32]を参考に、RIベースの新しいプログラムFMO-Xの開発を別途始めています。「富岳NEXT」上では、FMO-MP2レベルまではFMO-Xが主に担っている想定です。
開殻系と多参照系の扱いはVer. 3系で提供する予定です。2025年度の段階ではROHF計算の基幹部が完成し、スピン適合型CASSCFのプロトタイプの動作が確保できています[7]。相関エネルギーの補正は、ROHFにRMP2、CASSCFにNEVPT2として実装の準備を進めています。
[28] “Efficient recursive computation of molecular integrals over Cartesian Gaussian functions”, S. Obara, A. Saika, J. Chem. Phys., 84 (1986) 3963-3974.
[29] “フラグメント分子軌道法プログラムABINIT-MPのGPU による高速化”, 中野達也, 坂倉耕太, 成瀬彰, 片桐孝洋, 星野哲也, 大島聡史, 中島研吾, 石川岳志, 望月祐志, RIST NEWS, 71 (2025) 32-43.
[30] “Acceleration of fragment molecular orbital calculations with Cholesky decomposition approach”, Y. Okiyama, T. Nakano, K. Yamashita, Y. Mochizuki, N. Taguchi, S. Tanaka, Chem. Phys. Lett., 490 (2010) 84-89.
[31] “Fragment molecular orbital calculation using the RI-MP2 method”, T. Ishikawa, K. Kuwata, Chem. Phys. Lett., 474 (2009) 195-198.
[32] “RI-MP2 Gradient Calculation of Large Molecules Using the Fragment Molecular Orbital Method”, T. Ishikawa, K. Kuwata, J. Phys. Chem. Lett., 3 (2012) 375-379.
謝辞
ABINIT-MPの研究開発は、既述のようにJHPCNの2021年度からの課題プロジェクト[8]で続けられています(2026年度も採択済)。この中で、片桐孝洋先生と星野哲也先生(名古屋大学 情報基盤センター)、大島聡史先生(九州大学 情報基盤研究開発センター)にはHPCの専門的なアドバイスをいただいています。SX-AT関係では、滝沢寛之先生(東北大学 サイバーサイエンスセンター)にもご支援いただいています。
Ver. 2 Rev. 12向けの「富岳」での大規模なテスト計算は、感染症対策関係のHPCI課題{hp230017, hp240030, hp240526, hp250014}(代表者:望月祐志)の中で行ってきました。また、小規模FMOジョブを大量に実行して粗視化シミュレーションの一種である散逸粒子動力学(DPD)のパラメータを決めるアプローチ[33,34]を「富岳」上で推進する課題{hp230016, hp240013, hp250009}(代表者:望月祐志)とも連動しており、(株)JSOLの小沢拓部長には運営等でご協力いただいています。
FMODD関係では、福澤薫先生(大阪大学 大学院薬学研究科)、田中成典先生(神戸大学 分子フォトサイエンス研究センター)、加藤幸一郎先生(九州大学 大学院工学研究院)と連携しています。
SX-AT向けのベクトル化による高速化チューニング、ならびに解析機能の向上については長年NEC様からご支援をいただいており、実作業は加藤季広氏(NEC)と坂倉氏が担っています。また、複数の企業様からの立教大学へのご寄付や共同研究資金を得まして、Ver. 2系ABINIT-MPプログラムの研究開発と維持ができています。
RI関係では、PAICSの作者である石川岳志先生(鹿児島大学 学術研究院理工学域工学系)にご教示いただくこともあります。
HPCI拠点へのABINIT-MPのライブラリ整備やハンズオンセミナーでは、(一財)高度情報科学技術研究機構(RIST)に継続的にお世話になっています。
最後に、「計算工学ナビ」サイトでのABINIT-MP関係の情報公開で長年ご支援いただいている東京大学生産技術研究所の革新的シミュレーション研究センターに謝意を表します。
[33] “Fragment Molecular Orbital-based Parameterization Procedure for Mesoscopic Structure Prediction of Polymeric Materials”, K. Okuwaki, Y. Mochizuki, H. Doi, T. Ozawa, J. Phys. Chem. B, 122 (2018) 338-347.
[34] “フラグメント分子軌道(FMO)法を用いた散逸粒子動力学シミュレーションのための有効相互作用パラメータ算出の自動化フレームワーク”, 奥脇弘次, 土居英男, 望月祐志, J. Comp. Chem. Jpn., 17 (2018) 102-109.
コンタクト
ABINIT-MPのOpenシリーズのご利用(Pythonスクリプト含む)にご関心のある方は、取り纏め責任者の立教大学の望月祐志(fullmoon -at- rikkyo.ac.jp)にメールにてご連絡いただければ適宜対応させていただきます(-at-を@に変換してください)。